
Thalassemia: Insights into Genetic Blood Disorders
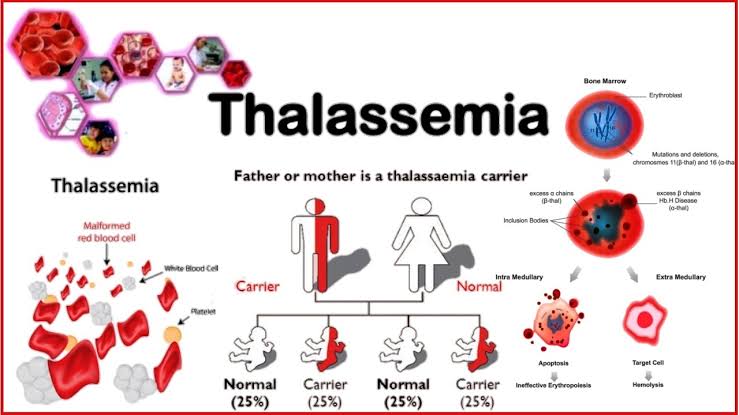
Thalassemia is a group of inherited blood disorders characterized by the body’s inability to produce hemoglobin properly. Hemoglobin is a protein in red blood cells responsible for carrying oxygen throughout the body. The primary issue in thalassemia is a defect in the production of either alpha or beta globin chains, which are crucial components of hemoglobin.
Types of Thalassemia
Thalassemia is a genetic blood disorder characterized by abnormal hemoglobin production. It’s primarily categorized into two main types based on which part of the hemoglobin molecule is affected:
1. Alpha Thalassemia
Alpha Thalassemia involves the alpha-globin chains of hemoglobin. The alpha-globin gene is located on chromosome 16, and each person has four copies of this gene (two from each parent). The severity of alpha thalassemia depends on the number of affected alpha-globin genes.
- Alpha Thalassemia Minor (Alpha Thalassemia Trait):
- Genetic Basis: Involves one or two mutated or deleted alpha-globin genes.
- Symptoms: Often asymptomatic or may present with mild anemia.
- Diagnosis: Identified through blood tests showing microcytic anemia and genetic testing.
- Alpha Thalassemia Major (Hemoglobin H Disease):
- Genetic Basis: Involves three out of the four alpha-globin genes being affected.
- Symptoms: Moderate to severe anemia, fatigue, jaundice, and an enlarged spleen. Individuals may also experience bone deformities and growth delays.
- Diagnosis: Blood tests showing the presence of Hemoglobin H (an abnormal form of hemoglobin) and genetic testing.
- Alpha Thalassemia with Hemoglobin Bart’s Hydrops Fetalis:
- Genetic Basis: Involves all four alpha-globin genes being affected.
- Symptoms: Extremely severe anemia and hydrops fetalis (a condition where there is an abnormal accumulation of fluid in the fetus). This condition is usually fatal before or shortly after birth.
- Diagnosis: Often diagnosed prenatally through ultrasound and genetic testing.
2. Beta Thalassemia
Beta Thalassemia involves the beta-globin chains of hemoglobin. The beta-globin gene is located on chromosome 11, and each person has two copies of this gene (one from each parent). The severity of beta thalassemia depends on the mutations in these genes.
- Beta Thalassemia Minor (Beta Thalassemia Trait):
- Genetic Basis: Involves one mutated beta-globin gene.
- Symptoms: Typically mild anemia, often asymptomatic or with minor symptoms. It’s usually discovered during routine blood tests.
- Diagnosis: Identified through blood tests showing microcytic anemia and genetic testing.
- Beta Thalassemia Intermedia:
- Genetic Basis: Involves two mutated beta-globin genes but with less severe mutations than beta thalassemia major.
- Symptoms: Moderate anemia and related symptoms like fatigue, but not as severe as beta thalassemia major. Symptoms can vary widely among individuals.
- Diagnosis: Similar to beta thalassemia major, but with less severe clinical features and blood test results.
- Beta Thalassemia Major (Cooley’s Anemia):
- Genetic Basis: Involves two severely mutated beta-globin genes.
- Symptoms: Severe anemia, growth delays, bone deformities, and complications such as an enlarged spleen and liver. Regular blood transfusions are often required to manage the condition.
- Diagnosis: Identified through blood tests showing very low levels of hemoglobin and increased fetal hemoglobin (HbF), along with genetic testing.
Summary
The classification of thalassemia into alpha and beta types helps guide diagnosis, treatment, and management strategies. Each type can range from mild to severe, depending on the number and nature of the gene mutations involved.
Symptoms
The symptoms of thalassemia vary based on the type and severity of the condition. Here’s a detailed look at the symptoms for each type:
1. Alpha Thalassemia
- Alpha Thalassemia Minor (Alpha Thalassemia Trait)
- Symptoms: Often asymptomatic or mildly symptomatic. Some individuals might experience mild anemia, fatigue, or pallor, but these symptoms are usually not severe and might not be noticeable without blood tests.
- Alpha Thalassemia Major (Hemoglobin H Disease)
- Symptoms:
- Anemia: Moderate to severe anemia with symptoms such as fatigue, weakness, and pallor.
- Jaundice: Yellowing of the skin and eyes due to increased breakdown of red blood cells.
- Enlarged Spleen and Liver: Can cause abdominal discomfort and swelling.
- Bone Deformities: Especially in the face and skull, due to marrow expansion in response to anemia.
- Growth Delays: Children may experience slower growth and development.
- Fatigue: Persistent tiredness and general weakness.
- Alpha Thalassemia with Hemoglobin Bart’s Hydrops Fetalis
- Symptoms:
- Severe Anemia: Extremely low levels of red blood cells, leading to severe fatigue.
- Hydrops Fetalis: Fluid accumulation in the fetus, leading to swelling and edema. This condition is typically fatal before or shortly after birth.
- No Survival Beyond Birth: This form is usually diagnosed through prenatal imaging and testing, and it often results in stillbirth or early neonatal death.
2. Beta Thalassemia
- Beta Thalassemia Minor (Beta Thalassemia Trait)
- Symptoms: Generally mild or absent. Individuals might experience mild anemia with symptoms like:
- Fatigue: Slight tiredness or weakness.
- Pallor: Pale skin.
- No significant complications are typically associated with this form.
- Beta Thalassemia Intermedia
- Symptoms: Moderate anemia, which can lead to:
- Fatigue: Persistent tiredness and weakness.
- Pallor: Pale skin.
- Enlarged Spleen and Liver: Abdominal discomfort and swelling.
- Bone Deformities: Less pronounced than in beta thalassemia major, but can occur.
- Growth Delays: Slower growth in children.
- Jaundice: Mild yellowing of the skin and eyes.
- Beta Thalassemia Major (Cooley’s Anemia)
- Symptoms: Severe and debilitating, including:
- Severe Anemia: Leading to extreme fatigue, weakness, and pallor.
- Frequent Infections: Due to compromised immune function and spleen involvement.
- Enlarged Spleen and Liver: Causing abdominal pain and swelling.
- Bone Deformities: Significant changes in facial bones and skull due to marrow expansion.
- Growth Delays: Noticeable delays in physical growth and development in children.
- Jaundice: Pronounced yellowing of the skin and eyes.
- Iron Overload: Resulting from repeated blood transfusions, leading to complications such as diabetes, heart disease, and liver damage. This is managed with iron chelation therapy.
General Symptoms Across Thalassemia Types
- Fatigue and Weakness: Common in all forms due to anemia.
- Pallor: Pale appearance of the skin.
- Enlarged Spleen and Liver: Can occur in more severe forms, leading to abdominal discomfort.
- Jaundice: May occur in moderate to severe cases due to increased red blood cell breakdown.
The severity and combination of symptoms can vary greatly among individuals, and the condition often requires regular monitoring and management to address these symptoms effectively.
Diagnosis
Diagnosing thalassemia involves several steps, including medical history evaluation, physical examination, and a series of specialized tests. Here’s a detailed look at the diagnostic process:
1. Medical History and Physical Examination
- Family History: Understanding the family history of thalassemia or other genetic disorders can provide important clues.
- Physical Examination: A healthcare provider may look for signs such as pallor, jaundice, or an enlarged spleen or liver.
2. Blood Tests
Several blood tests are crucial in diagnosing thalassemia:
- Complete Blood Count (CBC): Measures levels of red blood cells, hemoglobin, and hematocrit. In thalassemia, the red blood cells are often smaller (microcytic) and paler (hypochromic).
- Hemoglobin Electrophoresis: This test separates different types of hemoglobin to identify abnormal types. For example:
- Alpha Thalassemia: May show increased levels of Hemoglobin H (an abnormal type of hemoglobin).
- Beta Thalassemia: May show elevated levels of fetal hemoglobin (HbF) and, in some cases, Hemoglobin A2.
- Reticulocyte Count: Measures the number of immature red blood cells. Increased levels might indicate a response to anemia.
- Iron Studies: Includes tests like serum ferritin, transferrin saturation, and total iron-binding capacity (TIBC) to distinguish thalassemia from iron deficiency anemia.
3. Genetic Testing
- DNA Analysis: Identifies mutations in the alpha-globin or beta-globin genes. This is particularly useful for confirming a diagnosis of thalassemia and for prenatal diagnosis.
- Prenatal Testing: For expecting parents at risk, tests like chorionic villus sampling (CVS) or amniocentesis can detect thalassemia genes in the fetus.
4. Additional Tests
- Bone Marrow Biopsy: In rare cases, a bone marrow biopsy might be performed to assess the production of red blood cells, especially if there is uncertainty about the diagnosis.
- Ultrasound: Prenatal ultrasound can detect signs of severe thalassemia in the fetus, such as hydrops fetalis.
5. Differential Diagnosis
It’s important to differentiate thalassemia from other conditions that cause anemia, such as:
- Iron Deficiency Anemia: Distinguished through iron studies and response to iron supplementation.
- Sideroblastic Anemia: Identified by the presence of ringed sideroblasts in the bone marrow.
- Anemia of Chronic Disease: Assessed through clinical history and additional tests to identify underlying conditions.
Summary
The diagnostic approach for thalassemia combines clinical evaluation with specialized blood and genetic tests. Accurate diagnosis is essential for appropriate management and treatment planning. If thalassemia is suspected, it’s important to consult a hematologist or a specialist in genetic disorders for comprehensive evaluation and diagnosis.
Treatment
The treatment for thalassemia varies depending on the type and severity of the condition. Here’s a detailed overview of the common treatments and management strategies for different forms of thalassemia:
1. Alpha Thalassemia
- Alpha Thalassemia Minor (Alpha Thalassemia Trait)
- Treatment: Often requires no specific treatment. Regular monitoring may be sufficient, and individuals might only need periodic blood tests.
- Alpha Thalassemia Major (Hemoglobin H Disease)
- Treatment:
- Regular Blood Transfusions: To manage moderate to severe anemia. The frequency of transfusions varies depending on the individual’s needs.
- Iron Chelation Therapy: To manage iron overload from frequent blood transfusions. Medications like deferoxamine, deferasirox, or deferiprone are used to remove excess iron from the body.
- Folic Acid Supplements: To support red blood cell production.
- Monitoring and Management: Regular check-ups to monitor anemia, iron levels, and potential complications such as splenomegaly.
- Alpha Thalassemia with Hemoglobin Bart’s Hydrops Fetalis
- Treatment: This condition is typically fatal before or shortly after birth. Prenatal care and counseling are focused on managing the pregnancy and preparing for outcomes, including possible delivery plans.
2. Beta Thalassemia
- Beta Thalassemia Minor (Beta Thalassemia Trait)
- Treatment: Usually no treatment is required. Individuals may need periodic monitoring to ensure anemia remains mild.
- Beta Thalassemia Intermedia
- Treatment:
- Blood Transfusions: May be needed occasionally to manage anemia, but less frequently than in beta thalassemia major.
- Iron Chelation Therapy: Used as needed to prevent iron overload from occasional transfusions.
- Folic Acid Supplements: To help with red blood cell production.
- Monitoring: Regular follow-up to manage anemia, assess for complications, and adjust treatment as needed.
- Beta Thalassemia Major (Cooley’s Anemia)
- Treatment:
- Regular Blood Transfusions: Essential to manage severe anemia and maintain normal hemoglobin levels. Transfusions are typically needed every few weeks.
- Iron Chelation Therapy: Critical for managing iron overload due to frequent blood transfusions. Medications like deferoxamine, deferasirox, or deferiprone are used.
- Bone Marrow Transplant: May be a curative option, especially for children, if a suitable donor is available. This involves replacing the patient’s bone marrow with that of a healthy donor.
- Gene Therapy: Emerging treatments involving gene editing to correct the underlying genetic defect are being researched and tested.
- Monitoring and Supportive Care: Regular assessments to manage anemia, iron overload, and related complications. Includes monitoring for infections, managing growth delays, and addressing bone deformities.
General Supportive Measures
- Diet and Nutrition: Ensuring adequate nutrition to support overall health and address specific needs related to anemia and iron overload.
- Psychosocial Support: Psychological support and counseling for coping with the chronic nature of the disease and its impact on quality of life.
- Education and Counseling: For patients and families to understand the condition, treatment options, and management strategies.
Emerging and Experimental Treatments
- Gene Therapy: Research into gene therapy aims to correct the genetic mutations responsible for thalassemia. While still experimental, it holds promise for potentially curing or significantly improving the condition.
- New Drugs and Therapies: Ongoing research is exploring new medications and treatment approaches to improve outcomes and reduce complications.
Summary
Treatment for thalassemia is tailored to the specific type and severity of the disease, focusing on managing symptoms, preventing complications, and improving quality of life. Regular follow-up with a hematologist and a comprehensive care team is essential for effective management and treatment planning.
Management and Support
Managing thalassemia involves not only treating the symptoms but also providing ongoing support to improve quality of life and prevent complications. Here’s a comprehensive overview of management strategies and support for individuals with thalassemia:
1. Regular Monitoring and Medical Care
- Routine Check-Ups: Regular visits to a hematologist or a specialist to monitor the condition, assess treatment efficacy, and manage complications.
- Blood Tests: Frequent blood tests to monitor hemoglobin levels, iron levels, and overall health. This includes CBC, iron studies, and hemoglobin electrophoresis.
2. Treatment Adherence
- Medication Management: Adherence to prescribed medications, including iron chelation therapy and folic acid supplements, to manage anemia and prevent complications.
- Blood Transfusions: Following a scheduled transfusion plan to maintain adequate hemoglobin levels and avoid anemia-related complications.
3. Iron Overload Management
- Iron Chelation Therapy: Using medications like deferoxamine, deferasirox, or deferiprone to remove excess iron from the body due to frequent blood transfusions. Regular monitoring of iron levels is essential to adjust treatment as needed.
4. Lifestyle and Nutrition
- Balanced Diet: Eating a nutritious diet to support overall health and address specific needs. This might include foods rich in vitamins and minerals to help with anemia and overall well-being.
- Avoiding Excess Iron: Being cautious with iron-rich foods and supplements, as individuals with thalassemia may have excess iron in their bodies.
5. Psychological and Emotional Support
- Counseling: Access to mental health professionals to help individuals and families cope with the chronic nature of the condition and the stress associated with ongoing treatment.
- Support Groups: Joining support groups for people with thalassemia can provide emotional support, practical advice, and a sense of community.
6. Educational and Family Support
- Genetic Counseling: For families with a history of thalassemia, genetic counseling can help understand the risks for future pregnancies and provide information on inheritance patterns.
- Patient Education: Educating patients and families about thalassemia, treatment options, and self-care strategies. This includes understanding symptoms, managing side effects, and recognizing signs of complications.
7. Managing Complications
- Spleen and Liver Management: Regular monitoring and management of splenomegaly (enlarged spleen) and hepatomegaly (enlarged liver) to prevent complications and address discomfort.
- Bone Health: Monitoring and managing bone health issues, such as bone deformities and osteoporosis, which can arise from thalassemia and its treatments.
- Growth and Development: For children with thalassemia, monitoring growth and development to ensure they meet age-appropriate milestones and addressing any delays.
8. Specialized Care
- Bone Marrow Transplant: For eligible patients, a bone marrow transplant may be considered as a curative option. This involves finding a suitable donor and undergoing a complex procedure with associated risks.
- Gene Therapy: Experimental treatments involving gene therapy are being researched and may become available in the future. Keeping informed about advancements can be beneficial.
9. Lifestyle Adjustments
- Activity Level: Adjusting physical activity based on energy levels and overall health. Encouraging regular but manageable exercise can help maintain physical fitness and well-being.
- Travel and Lifestyle: Planning for travel and daily activities while managing thalassemia, including ensuring access to medical care and medications when away from home.
10. Emergency Preparedness
- Emergency Plan: Having a plan in place for managing potential emergencies, such as severe anemia or complications, including knowing when to seek urgent medical care and having access to emergency contact information.
Summary
Effective management of thalassemia requires a multi-faceted approach that includes regular medical care, adherence to treatment, lifestyle adjustments, and emotional support. Working closely with healthcare providers and utilizing available resources can significantly improve outcomes and quality of life for individuals with thalassemia.
Prevention and Research
Prevention
Preventing thalassemia focuses primarily on reducing the incidence of the disease through genetic screening and counseling, as well as raising awareness. Here’s how prevention strategies are typically approached:
1. Genetic Screening and Counseling
- Carrier Screening: Testing individuals, particularly those with a family history of thalassemia or who belong to ethnic groups at higher risk (e.g., Mediterranean, Middle Eastern, Southeast Asian), to determine if they carry a thalassemia gene mutation.
- Preconception Counseling: For couples planning to have children, genetic counseling can provide information about the risks of passing thalassemia genes to offspring and discuss options.
- Prenatal Testing: Offering testing for expectant parents who are known carriers. Methods include:
- Chorionic Villus Sampling (CVS): Performed between the 10th and 13th weeks of pregnancy to test for thalassemia genes in the placenta.
- Amniocentesis: Performed between the 15th and 20th weeks of pregnancy to test the amniotic fluid for genetic mutations.
2. Public Awareness and Education
- Awareness Programs: Educating communities about thalassemia, its genetic basis, and the importance of carrier screening.
- Educational Materials: Providing information through pamphlets, websites, and community health programs to increase awareness and understanding of the disease.
3. Carrier Prevention Programs
- Informed Decision-Making: Ensuring that individuals who are carriers have access to information about their reproductive options, including the potential use of assisted reproductive technologies like preimplantation genetic diagnosis (PGD).
Research
Ongoing research aims to improve the understanding, treatment, and management of thalassemia. Key areas of focus include:
1. Gene Therapy
- Research and Development: Investigating ways to correct the genetic mutations responsible for thalassemia. This includes:
- Gene Editing: Techniques like CRISPR/Cas9 to correct defective genes in stem cells.
- Gene Addition: Adding functional copies of the beta-globin gene into patient cells.
2. New Treatments
- Novel Medications: Developing new drugs to manage symptoms and reduce complications. This includes research into:
- Fetal Hemoglobin Inducers: Medications that increase the production of fetal hemoglobin (HbF), which can partially compensate for defective hemoglobin.
- Improved Iron Chelators: Developing new chelation therapies with fewer side effects and better efficacy.
3. Bone Marrow and Stem Cell Transplants
- Improving Outcomes: Enhancing the success rates of bone marrow and stem cell transplants, including better matching techniques and reducing the risk of graft-versus-host disease (GVHD).
4. Quality of Life and Supportive Care
- Managing Complications: Research into better ways to manage complications associated with thalassemia, such as iron overload and bone deformities.
- Psychosocial Support: Studying the impact of thalassemia on mental health and developing interventions to support emotional well-being.
5. Genetic Research
- Understanding Variants: Investigating the various genetic mutations that cause thalassemia to improve diagnosis and treatment.
- Population Studies: Analyzing genetic data from different populations to understand the distribution and prevalence of thalassemia genes.
6. Global Health Initiatives
- International Collaboration: Working with global health organizations to improve screening programs, access to care, and treatment in regions where thalassemia is more prevalent.
- Data Collection: Gathering and analyzing data to inform public health policies and improve disease management strategies worldwide.
Summary
Preventing thalassemia involves proactive genetic screening, counseling, and public education to reduce the incidence and impact of the disease. Research continues to advance the understanding and treatment of thalassemia, focusing on gene therapy, novel treatments, and improving patient care. Ongoing studies and clinical trials aim to provide better outcomes and quality of life for individuals affected by thalassemia.
Thalassemia is a significant public health issue in India, where it is prevalent due to the high frequency of the genetic mutations associated with the condition. The rate of thalassemia in India varies regionally, with certain areas showing higher prevalence due to genetic factors. Here are key points about the prevalence and impact of thalassemia in India:
Prevalence
- Carrier Frequency: The carrier rate of thalassemia in India varies widely across different regions. For example:
- Beta Thalassemia: The carrier rate is particularly high in states like Punjab, Gujarat, Maharashtra, and parts of Uttar Pradesh. In some of these areas, carrier rates can be as high as 3-10% of the population.
- Alpha Thalassemia: Generally found in higher frequencies in southern and northeastern states such as Tamil Nadu, Kerala, and Assam.
- Disease Burden: It is estimated that approximately 10,000 to 15,000 new cases of thalassemia major are diagnosed each year in India. The overall prevalence of thalassemia major is estimated to be around 1 in 10,000 live births, though this can vary.
Impact
- High Incidence in Certain Regions: Thalassemia is more common in regions with higher carrier rates. For example, in Punjab, one of the most affected states, there is a high incidence of thalassemia major and minor.
- Healthcare Challenges: Managing thalassemia involves significant healthcare resources, including regular blood transfusions and iron chelation therapy. In India, access to these treatments can be inconsistent, particularly in rural or less-developed areas.
